撰稿 / 張鳳吟 (科學推展中心特約編輯)
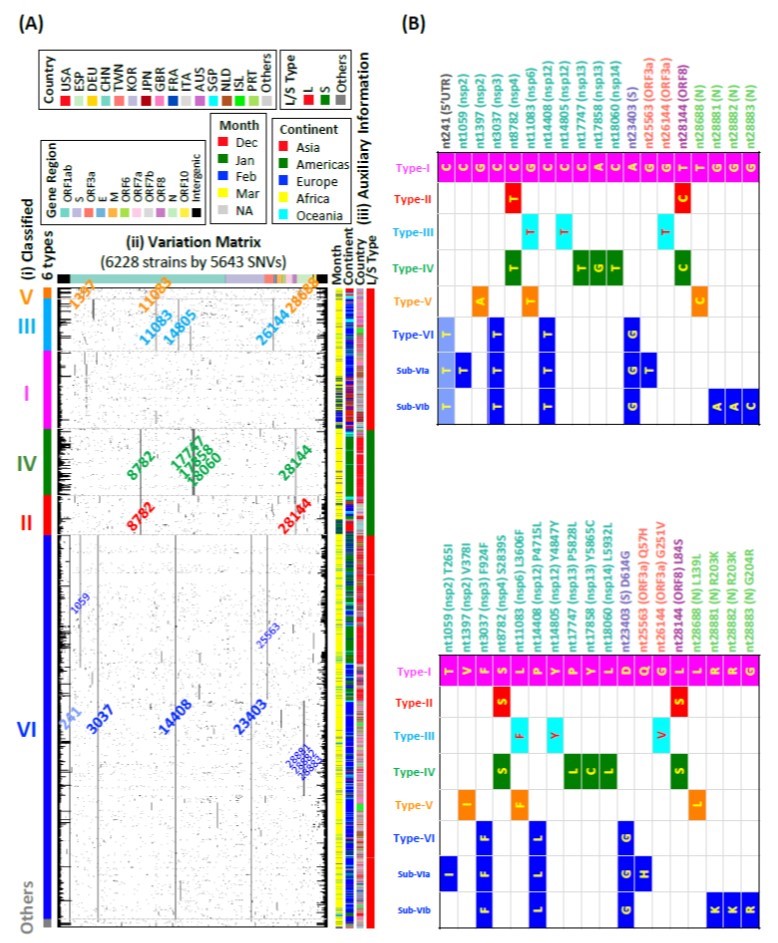
圖1、 (a)是進一步分析2020年4月下載的6,228個病毒株數據呈現的病毒變異矩陣(6228病毒株×5643位點),其中矩陣上方不同顏色的橫條表示病毒基因體不同的蛋白區段,左方羅馬數字代表病毒株分型,中間阿拉伯數字為發生變異的位置。 (b) 病毒六大型和第六型子型的特徵變異的註解,上圖為變異核苷酸註解,下圖為變異胺基酸註解。
新冠肺炎(COVID-19)自第一例人傳人於2019年12月在中國武漢被揭露後,迄今已遍布世界五大洲,逾兩億人感染,超過450萬人死亡,且病毒持續變異中,使得疫苗與藥物研發更添變數。在疫情爆發之初,中研院廖俊智院長旋即帶領中研院統計所與生物多樣性中心團隊展開新冠病毒研究,以大數據與統計的方式分析病毒變異株,成績斐然,成果除發表於《美國國家科學院院刊》(PNAS)[1],並持續追蹤監測新冠病毒的變異趨勢,有助於了解病毒傳播發展及疫苗效率的評估。
新冠肺炎的病毒SARS-CoV-2為一單股正鏈RNA,其基因體長約3萬個核苷酸(nucleotides),以A、U (T)、C、G不同鹼基組成。除了頭尾兩個區段序列(5’、3’非轉譯區),中間基因體包含了11個解碼區:刺突蛋白(S)、包膜蛋白(E)、跨膜蛋白(M)、核殼蛋白(N),以及數個開放閱讀框架(open reading frames,ORFs)。每一段有不同長度與功能,其中刺突蛋白會和人體細胞上的第二型血管收縮素轉化酶(ACE2)結合,是病毒進入人體的鑰匙。由於RNA只有單股,相較於穩定的雙股的DNA,RNA相當不穩定,當病毒進入細胞後,在不斷複製基因的過程容易出錯,每個錯誤,就會讓核苷酸位點發生突變。有些單一核苷酸變異(single nucleotide variations,SNVs)可能會增加病毒適應環境的能力,加速傳播效率並影響疫苗效價。
2008年建立的全球共享流感數據倡議組織GISAID是目前全球最大的流感及新冠病毒數據平台,中研院團隊2020年3月從GISAID先取得1,932筆病毒株基因序列資料,以完整基因體進行統計群集分析與親緣關係樹(呈現個體之間親緣關係的樹狀圖)分析,五種樹狀圖分辨出類似的病毒株類型,研究人員進一步將每個病毒株基因體和武漢原始株比對,標記突變的部分,依據樹狀圖分支排序成矩陣圖(圖1a)。
從圖1結果,研究人員定義出病毒可分為六大型(I~VI),並發現這六型可由14個單一核苷酸變異(SNVs)來決定,扣掉位於5’非轉譯區的SNV C241T(代表241位置的C變異為T),每一型(除了第一型)都能用剩下的13個SNVs中的至少2個來定義。這個結果在後續更多樣本數的兩組數據(6,228、38,248病毒株)得到相同的證實。因此研究團隊提出,病毒株可以用此14個SNVs印記來分類,不需要參考完整序列的親緣關係。建立在這個基礎上,研究人員更進一步開發兩種演算法:一對一序列排比(pairwise sequence alignment)與文字探勘(text mining),在分析大筆數據時,能大幅增加運算效率,且正確率在98%以上。
從2020年3、4月開始,第六型病毒株逐漸成為全世界主流的病毒株(圖2)。第六型是由C241T、C3037T、C14408T、A23403G(刺突蛋白S)四個變異所定義。研究人員發現,疫情流行期間,偶而會出現只有四個變異中的其中幾個變異的變異株,但它們都沒有傳播很久,只有在四個特徵變異同時出現時,變異株才能維持,暗示這四個位置的同時變異,可能讓病毒對環境有更強的適應力。同樣情形也發生在其他許多國家。現在第六型的病毒持續在全球肆虐,目前傳染力極強的Alpha、Beta、Gamma、Delta都是第六型的子型,中研院持續監控既有病毒株與新興變異株中。
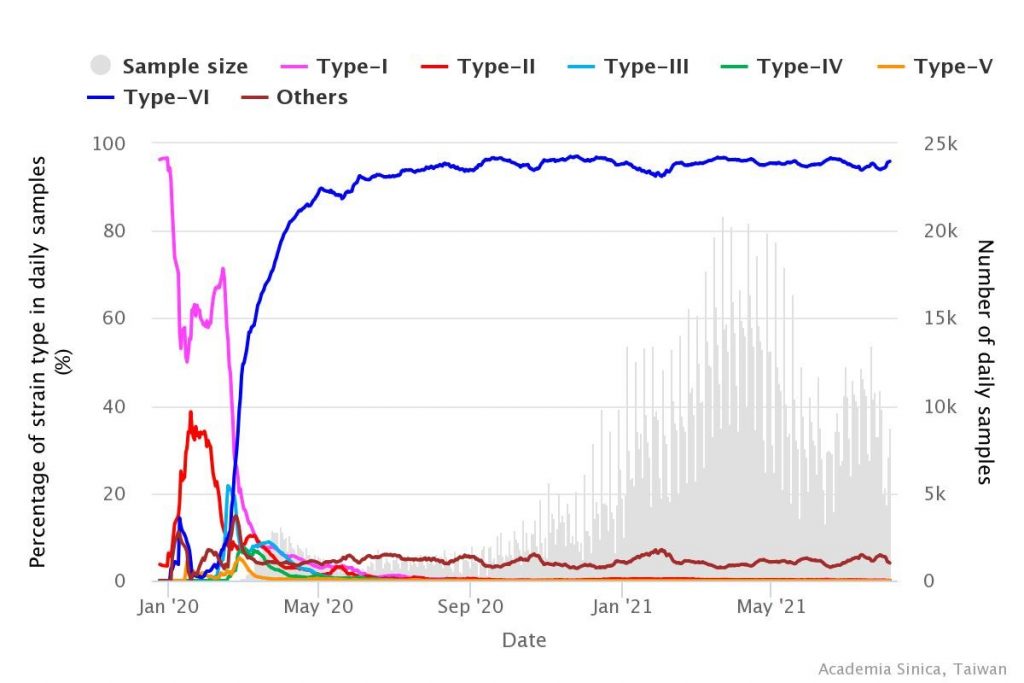
圖2、全球6大型新冠病毒隨時間的消長 (截至2021/9/10) (來源:中研院病毒變異全球即時監測網)
為因應病毒的持續變異,中央研究院院長廖俊智博士帶領院內各領域專家組成病毒即時監控跨領域團隊,團隊成員包括中研院統計科學研究所研究員楊欣洲、中研院統計科學研究所研究員陳君厚、生物醫學科學研究所研究員林宜玲、生物醫學科學研究所研究員陶秘華、生物化學研究所副研究員徐尚德。並以此成果為依據,由中研院資訊科學研究所研究員陳伶志架設「病毒變異全球即時監測網」[2],除了掌握病毒分型的趨勢,也能即時監測世界各國正在發生的新興病毒變異,提供未來病毒傳播與疫苗有效的評估。
參考文獻
[1] Yang, H.-C.*, Chen, C.-h., Wang, J.-H., Liao, H.-C., Yang, C.-T., Chen, C.-W., Lin, Y.-C., Kao, C.-H., Lu, M.-Y. J. and Liao, J. C.* , Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proceedings of the National Academy of Sciences of the United States of America 117, 48
[2] 病毒變異全球即時監測網網址:https://sarscov2.sinica.edu.tw/